Written in collaboration with Wendy Mendes.
Diabetes mellitus is disease that affects a large part of the world population, being frequently cited as a driving cause of premature death, as well as a risk factor for other pathologies, including pandemic diseases such as COVID-19.
When diabetes is the subject, insulin is almost automatically popped out in our minds. However, there is another important hormone that could be involved in some forms of this disease, the islet amyloid polypeptide, also known as amylin. This is a 37-residue peptide hormone co-secreted with insulin at a ratio of 1:100 by the pancreatic β-cells. Because diabetes is extensively related to glucose control mechanisms, this disease envolves the behavior of other associated hormones; and amylin plays a critical role in glucose homeostasis performing a synergistic activity with insulin.
Diabetes corresponds to a group of metabolic disorders, where type II diabetes is highlighted being the most frequent type. This form of the disease is a result of insufficient insulin production and/or resistance to insulin response, similar to other forms of the disease. The hallmark of type II diabetes is hyperglycemia resulting from disturbances in glucose processing, where amylin plays a critical role. This hormone has the ability to aggregate into fibrils that can generate amyloid deposits. Due to the cytotoxicity of this amyloid formation, amylin is related to the pathophysiology of type II diabetes, because the pancreatic β-cells are often destroyed by the amyloid formation.
Being a gene encoded hormone, amylin could suffer modifications driven by mutations on its gene. And such modifications could increase or decrease the amylin amyloid formation capacity, altering its biological activity. For instance, studies with amylin orthologues indicated that the rat amylin has a lower propensity for aggregation, and based on such modifications the drug pramlintide was developed. On the other hand, some mutations such as S20G, driven by a single nucleotide polymorphism (SNP) seem to be capable to increase this ability.
In fact, SNPs can lead to changes in the structure and/or function of a peptide. And the effects of these changes in amylin are still not fully understood, especially since it is a dynamic and flexible molecule, evaluating the effects of SNPs on the amylin sequence can be a challenge. Currently, computational tools, e.g. SNP effect predictors, molecular modeling and molecular dynamics, have been used to characterize and evaluate SNPs, in addition to generating new insights into their relationship with the most diverse diseases.
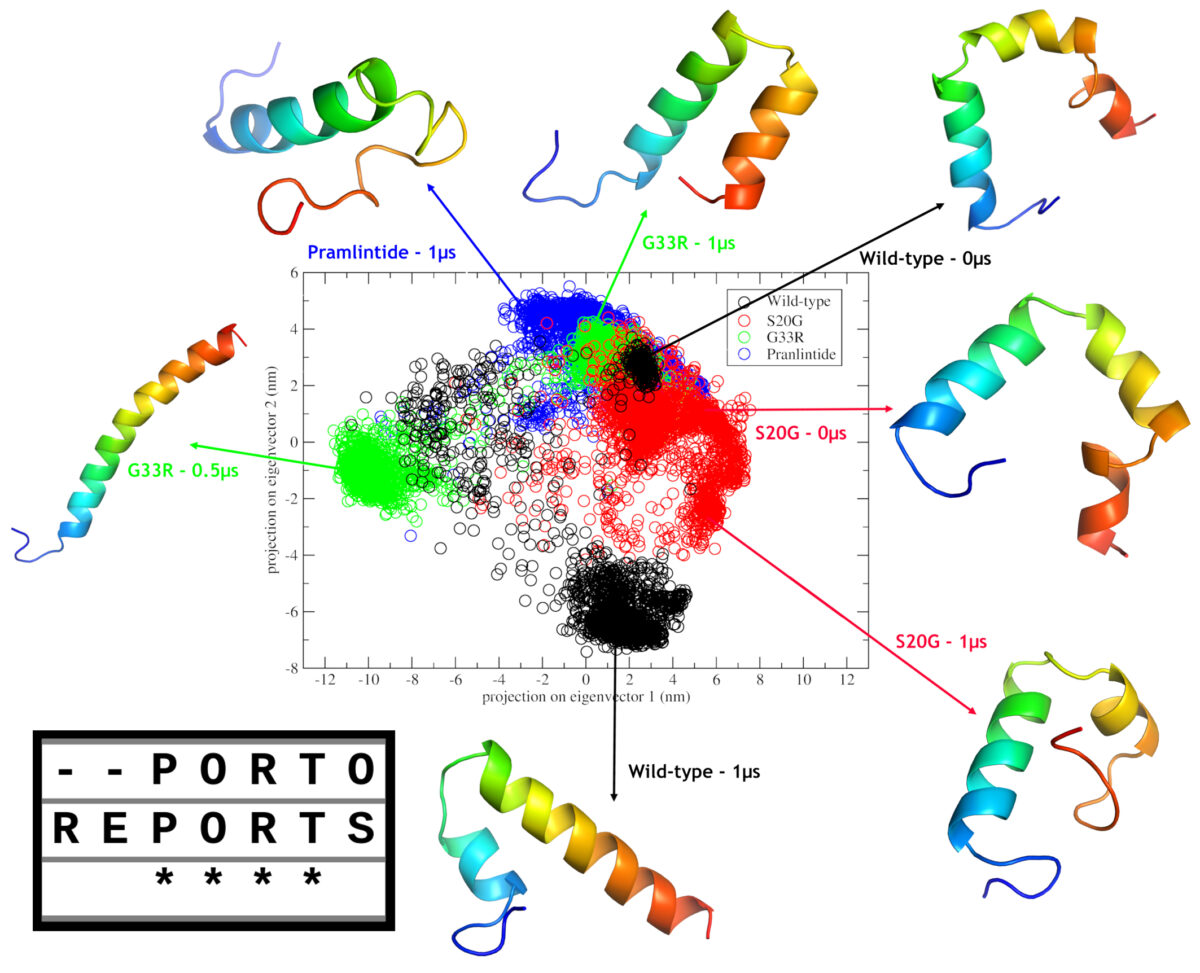
In this study by Mendes et al. (2021), two amylin SNPs (including S20G), where subjected to in silico analysis, in order to gain insights on their effects on the structure. Our results indicated that both mutations have aggregation potential and may cause changes in the monomeric forms when compared with wild-type amylin. In additon, when compared to pramlintide we could infer that second α-helix maintenance may be related to the aggregation potential.
The S20G mutation has a frequence >1% in east asian population, and this could be related to the fact that asian have more rates of type II diabetes. However the most intriguing mutation is G33R due two factors. Firstly, the introduction of an arginine at such position, made the C-Terminal cationic, which in turn, caused a repultion to N-Terminal, leading to a completely different structural type, a complete α-helix, which could be observed in the headline figure of this post; and secondly, this mutation was only observed in a single individual. Thus, we don’t know wheter this mutation is a de novo mutation or the frequence is very low due to the putative deleterious effects driven by this different structural type.
This study could help to better understand the impact of mutations on the wild-type amylin sequence, as a starting point for the evaluation and characterization of other variations. Moreover, these findings could improve the health of patients with type II diabetes with this genetic background.
Quality assessment:
Originality ☆☆☆☆✭
Rigor ☆☆☆☆✭
Significance to the field ☆☆☆☆☆
Interest to general audience ☆☆☆☆☆
Quality of writing ☆☆☆☆☆
Overall quality of the study ☆☆☆☆✭
Reference
Mendes et al. (2021) Structural effects driven by rare point mutations in amylin hormone, the type II diabetes-associated peptide. BBA General Subjects, vol. 1865, no. 8, 129935. https://doi.org/10.1016/j.bbagen.2021.129935